asyncmd.trajectory.SlurmTrajectoryFunctionWrapper#
The asyncmd.trajectory.SlurmTrajectoryFunctionWrapper can wrap any executable that works on the trajectories underlying the asyncmd.Trajectory objects. The wrapped executable is turned into an asynchronously callable subprocess and as for the functions in the PyTrajectoryFunctionWrapper, the results are cached for each trajectory such that reapplying a function is cheap. SlurmTrajectoryFunctionWrapper will submit the execution via SLURM and thereby enables the use of multiple cores distributed over multiple nodes when applying the same function to multiple different trajectories at once. The executable is expected to take the path to the trajectory and structure files on the command line, any user specified command line flags and arguments can also be passed though. Please have a look at the docstring of the SlurmTrajectoryFunctionWrapper for more.
We will use the SlurmTrajectoryFunctionWrapper here to wrap the state and descriptor functions for capped alanine dipeptide. The content of the file ala_cv_funcs.py is printed below. Please have a look at the functions we import to make sure you understand what they return. Being able to write functions that can be used on your molecular system of interest yourself is the key to use asyncmd to its full potential.
In addition to the state functions you should have a look at the code for parsing command line arguments. This is the magic bit that converts the python function into an executable. You could of course also use any other (non-python) executable that takes trajectory files on the command line as required. Additionally the executable either needs to write out its results as numpy npz file or you must define a custom loading function that will be called with a filename as argument and is expected to return a numpy array with the results. The custom loading function option enables you to write out the results to disk in any (custom) format as long as you can find or write a function to read it.
Imports#
%matplotlib inline
import asyncio
import matplotlib.pyplot as plt
import numpy as np
import asyncmd
from asyncmd import trajectory as asynctraj
Have a look at the function definitions#
# this is just to have a look at the file content
from pygments import highlight
from pygments.lexers import PythonLexer
from pygments.formatters import HtmlFormatter
import IPython
with open('../resources/ala_cv_funcs.py') as f:
code = f.read()
formatter = HtmlFormatter()
IPython.display.HTML('<style type="text/css">{}</style>{}'.format(
formatter.get_style_defs('.highlight'),
highlight(code, PythonLexer(), formatter)))
#!/usr/bin/env python3
"""
State und descriptor functions for capped alanine dipetide.
"""
import argparse
import numpy as np
import MDAnalysis as mda
from MDAnalysis.lib.distances import calc_dihedrals
def alpha_R(traj, skip=1):
"""
Calculate alpha_R state function.
The alpha_R state is defined in the space of the two dihedral angles psi
and phi, for a configuration to belong to the state:
phi: -pi < phi < 0
psi: -50 degree < psi < 30 degree
Parameters
----------
traj : asyncmd.Trajectory
The trajectory for which the state function is calculated.
skip : int, optional
stride for trajectory iteration, by default 1
Returns
-------
numpy.ndarray, shape=(n_frames,)
Array with boolean values for every configuration on the trajectory
indicating if a configuration falls into the state or not.
"""
u = mda.Universe(traj.structure_file, *traj.trajectory_files)
psi_ag = u.select_atoms("resname ALA and name N") # idx 6
psi_ag += u.select_atoms("resname ALA and name CA") # idx 8
psi_ag += u.select_atoms("resname ALA and name C") # idx 14
psi_ag += u.select_atoms("resname NME and name N") # idx 16
phi_ag = u.select_atoms("resname ACE and name C") # idx 4
phi_ag += u.select_atoms("resname ALA and name N") # idx 6
phi_ag += u.select_atoms("resname ALA and name CA") # idx 8
phi_ag += u.select_atoms("resname ALA and name C") # idx 14
# empty arrays to fill
state = np.full((len(u.trajectory[::skip]),), False, dtype=bool)
phi = np.empty((len(u.trajectory[::skip]),), dtype=np.float64)
psi = np.empty((len(u.trajectory[::skip]),), dtype=np.float64)
for f, ts in enumerate(u.trajectory[::skip]):
phi[f] = calc_dihedrals(*(at.position for at in phi_ag), box=ts.dimensions)
psi[f] = calc_dihedrals(*(at.position for at in psi_ag), box=ts.dimensions)
# make sure MDAnalysis closes the underlying trajectory files directly
u.trajectory.close()
# phi: -pi -> 0
# psi: > -50 but smaller 30 degree
deg = 180/np.pi
state[(phi <= 0) & (-50/deg <= psi) & (psi <= 30/deg)] = True
return state
def C7_eq(traj, skip=1):
"""
Calculate C7_eq state function.
The C7_eq state is defined in the space of the two dihedral angles psi
and phi, for a configuration to belong to the state:
phi: -pi < phi < 0
psi: 120 degree < psi < 200 degree
Parameters
----------
traj : asyncmd.Trajectory
The trajectory for which the state function is calculated.
skip : int, optional
stride for trajectory iteration, by default 1
Returns
-------
numpy.ndarray, shape=(n_frames,)
Array with boolean values for every configuration on the trajectory
indicating if a configuration falls into the state or not.
"""
u = mda.Universe(traj.structure_file, *traj.trajectory_files)
psi_ag = u.select_atoms("resname ALA and name N") # idx 6
psi_ag += u.select_atoms("resname ALA and name CA") # idx 8
psi_ag += u.select_atoms("resname ALA and name C") # idx 14
psi_ag += u.select_atoms("resname NME and name N") # idx 16
phi_ag = u.select_atoms("resname ACE and name C") # idx 4
phi_ag += u.select_atoms("resname ALA and name N") # idx 6
phi_ag += u.select_atoms("resname ALA and name CA") # idx 8
phi_ag += u.select_atoms("resname ALA and name C") # idx 14
# empty arrays to fill
state = np.full((len(u.trajectory[::skip]),), False, dtype=bool)
phi = np.empty((len(u.trajectory[::skip]),), dtype=np.float64)
psi = np.empty((len(u.trajectory[::skip]),), dtype=np.float64)
for f, ts in enumerate(u.trajectory[::skip]):
phi[f] = calc_dihedrals(*(at.position for at in phi_ag), box=ts.dimensions)
psi[f] = calc_dihedrals(*(at.position for at in psi_ag), box=ts.dimensions)
# make sure MDAnalysis closes the underlying trajectory files directly
u.trajectory.close()
# phi: -pi -> 0
# psi: 120 -> 200 degree
deg = 180/np.pi
state[(phi <= 0) & ((120/deg <= psi) | (-160/deg >= psi))] = True
return state
def descriptor_func_psi_phi(traj, skip=1):
"""
Calculate psi and phi angle internal coordiantes.
Parameters
----------
traj : asyncmd.Trajectory
Input trajectory.
skip : int, optional
stride for trajectory iteration, by default 1
Returns
-------
np.ndarray
psi, phi values for trajectory, shape=(n_frames, 2)
"""
u = mda.Universe(traj.structure_file, *traj.trajectory_files)
psi_ag = u.select_atoms("index 6 or index 8 or index 14 or index 16")
phi_ag = u.select_atoms("index 4 or index 6 or index 8 or index 14")
# empty arrays to fill
phi = np.empty((len(u.trajectory[::skip]), 1), dtype=np.float64)
psi = np.empty((len(u.trajectory[::skip]), 1), dtype=np.float64)
for f, ts in enumerate(u.trajectory[::skip]):
phi[f, 0] = calc_dihedrals(*(at.position for at in phi_ag), box=ts.dimensions)
psi[f, 0] = calc_dihedrals(*(at.position for at in psi_ag), box=ts.dimensions)
# make sure MDAnalysis closes the underlying trajectory files directly
u.trajectory.close()
return np.concatenate((psi, phi), axis=1)
if __name__ == "__main__":
parser = argparse.ArgumentParser(
description="Calculate CV values for alanine dipeptide",
)
parser.add_argument("structure_file", type=str)
parser.add_argument("trajectory_files", type=str, nargs="+")
parser.add_argument("output_file", type=str)
parser.add_argument("-f", "--function", type=str,
default="descriptors",
choices=["alphaR", "C7eq", "descriptors_psi_phi"])
parser.add_argument("-s", "--skip", type=int, default=1)
args = parser.parse_args()
# NOTE: since args is a namespace args.trajectory_file will be the path to
# the trajectory file, i.e. we can pass args instead of an
# aimmd.Trajectory to the functions above
if args.function == "descriptors_psi_phi":
vals = descriptor_func_psi_phi(args, skip=args.skip)
elif args.function == "alphaR":
vals = alpha_R(args, skip=args.skip)
elif args.function == "C7eq":
vals = C7_eq(args, skip=args.skip)
np.save(args.output_file, vals)
Create the wrapped state functions#
As you hopefully have guessed from the code above each state functions returns one value for every frame in the trajectory, i.e. their output is expected to be of shape (len(trajectory),) and the values simply indicate whether each frame is to be considered part of the respective state (True) or not (False).
When called via the command line the code will write out the results to args.output_file as numpy npz file. This is then automatically loaded by asyncmd and we do not have to define a custom loading function.
The script will be called via the sbatch_script, which is printed below. It is very minimal and depending on the configuration of the cluster you are running on, you probably want to add at least a partition to ensure that you are not blocking a whole node for the 1 core we are requesting to run the CV calculation. The string “{cmd_str}” will be replaced by asyncmd with the specific command to run the CV calculation for the respective trajectory.
from pygments.lexers import BashLexer
with open('../resources/ala_calc_cvs.slurm') as f:
code = f.read()
formatter = HtmlFormatter()
IPython.display.HTML('<style type="text/css">{}</style>{}'.format(
formatter.get_style_defs('.highlight'),
highlight(code, BashLexer(), formatter)))
#!/bin/bash -l
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --mem=4500
### Things you might want to set to run resource-efficient (non-exhaustive)
##SBATCH --partition=
##SBATCH --time=
##SBATCH --nodes=
# Note: make sure that you activate the correct environment, preferably the same you run asyncmd from
source ~/asyncmd_workshop_test/source_modules_phys.sh
srun {cmd_str}
C7_eq_wrapped = asynctraj.SlurmTrajectoryFunctionWrapper(executable="../resources/ala_cv_funcs.py",
sbatch_script="../resources/ala_calc_cvs.slurm",
call_kwargs={"--function": "C7eq",
"--skip": 1,
},
)
# the optional call_kwargs argument can be used to specify additional keyword arguments
# [we pass skip=1 which does not do anything because it is the default value only to show that call_kwargs exists]
alpha_R_wrapped = asynctraj.SlurmTrajectoryFunctionWrapper(executable="../resources/ala_cv_funcs.py",
sbatch_script="../resources/ala_calc_cvs.slurm",
call_kwargs={"--function": "alphaR",
"--skip": 1,
},
)
Load two different configurations as asyncmd.Trajectory#
They belong to two different states.
# create an asyncmd.Trajectory of the initial configuration from the `GmxEngine.ipynb` notebook
conf_in_alphaR = asyncmd.Trajectory(trajectory_files="../resources/gromacs/capped_alanine_dipeptide/conf_in_alphaR.trr",
structure_file="../resources/gromacs/capped_alanine_dipeptide/conf.gro",
)
# create a second asyncmd.Trajectory of another configuration (in another state)
conf_in_C7eq = asyncmd.Trajectory(trajectory_files="../resources/gromacs/capped_alanine_dipeptide/conf_in_C7eq.trr",
structure_file="../resources/gromacs/capped_alanine_dipeptide/conf.gro",
)
Apply the state functions to both configurations/trajectories simultaneously#
We use asyncio.gather as usual to collect multiple tasks/coroutine executions and execute them all at once via SLURM.
import time
states = [alpha_R_wrapped, C7_eq_wrapped]
start = time.time()
states_for_conf_in_alphaR = await asyncio.gather(*(sf(conf_in_alphaR) for sf in states))
states_for_conf_in_C7eq = await asyncio.gather(*(sf(conf_in_C7eq) for sf in states))
end = time.time()
print(f"States (alphaR, C7_eq) for conf_in_alphaR: {states_for_conf_in_alphaR}.")
print(f"States (alphaR, C7_eq) for other_conf: {states_for_conf_in_C7eq}.")
print(f"The calculation took {round(end-start, 4)} seconds")
States (alphaR, C7_eq) for conf_in_alphaR: [array([ True]), array([False])].
States (alphaR, C7_eq) for other_conf: [array([False]), array([ True])].
The calculation took 20.22 seconds
Applying the state functions again, we can observe that the result is obtained much faster because it has been cached#
The caching applies to all wrapped functions, i.e. the return values of all wrapped functions operating on asyncmd.Trajectory objects are cached. This means that all (potentially costly) functions operating on a trajectory will have to be evaluated only once (the first time they are called), even if called multiple places in the code (e.g. because the order of execution was yet undetermined).
Note that the comparison here where the trajectories just have one frame (and computing the values is cheap and fast) is one of the most unfavorable ones possible for the caching mechanism. However, the overhead of submitting and waiting for the slurm job works in our favor ;)
start = time.time()
states_for_conf_in_alphaR = await asyncio.gather(*(sf(conf_in_alphaR) for sf in states))
states_for_conf_in_C7eq = await asyncio.gather(*(sf(conf_in_C7eq) for sf in states))
end = time.time()
print(f"States (alphaR, C7_eq) for conf_in_alphaR: {states_for_conf_in_alphaR}.")
print(f"States (alphaR, C7_eq) for other_conf: {states_for_conf_in_C7eq}.")
print(f"The calculation took {round(end-start, 4)} seconds")
States (alphaR, C7_eq) for conf_in_alphaR: [array([ True]), array([False])].
States (alphaR, C7_eq) for other_conf: [array([False]), array([ True])].
The calculation took 0.0053 seconds
Note: The naming almost gave it away, each of the conformations is in one of the states.
Chain the application of TrajectoryFunctionWrappers#
Depending on your collective variable functions it can make sense to cache (computationally costly) intermediate results. An example would be two states that are defined in terms of the same descriptors. This is the case for the \(\alpha_R\) and \(C7_{eq}\) states, which are both defined in terms of the two dihedrals \(\psi\) and \(\phi\). We will demonstrate the chaining of TrajectoryFunctionWrappers here by reimplementing the two state functions alpha_R and C7_eq on top of the output of the wrapped function descriptor_func_psi_phi.
psi_phi_wrapped = asynctraj.SlurmTrajectoryFunctionWrapper(executable="../resources/ala_cv_funcs.py",
sbatch_script="../resources/ala_calc_cvs.slurm",
call_kwargs={"--function": "descriptors_psi_phi",
"--skip": 1,
},
)
(Re)define the state functions in \(\psi\) and \(\phi\)#
Using the two functions below, the (wrapped) descriptor_func_psi_phi will only be called once per trajectory even if we call both state functions. In addition we can also wrap the newly defined asynchronous functions to cache their results as usual by passing them to a PyTrajectoryFunctionWrapper.
async def alpha_R_redef(traj):
# phi: -pi < phi < 0
# psi: -50 degree < psi < 30 degree
psi_phi = await psi_phi_wrapped(traj)
psi, phi = psi_phi[:, 0], psi_phi[:, 1]
state = np.zeros((len(traj),), dtype=bool)
deg = 180/np.pi
state[(phi <= 0) & (-50/deg <= psi) & (psi <= 30/deg)] = True
return state
async def C7_eq_redef(traj):
# phi: -pi -> 0
# psi: 120 -> 200 degree
psi_phi = await psi_phi_wrapped(traj)
psi, phi = psi_phi[:, 0], psi_phi[:, 1]
state = np.zeros((len(traj),), dtype=bool)
deg = 180/np.pi
state[(phi <= 0) & ((120/deg <= psi) | (-160/deg >= psi))] = True
return state
# applying the second state function should be much faster since it will use the cached intermediate results for φ and ψ
confs = [conf_in_alphaR, conf_in_C7eq]
start = time.time()
results_alphaR_redef = await asyncio.gather(*(alpha_R_redef(conf) for conf in confs))
end = time.time()
print(f"alpha_R results for both configurations are: {results_alphaR_redef}.")
print(f"The calculation took {round(end-start, 4)} seconds")
start = time.time()
results_C7eq_redef = await asyncio.gather(*(C7_eq_redef(conf) for conf in confs))
end = time.time()
print(f"C7_eq results for both configurations are: {results_C7eq_redef}.")
print(f"The calculation took {round(end-start, 4)} seconds")
alpha_R results for both configurations are: [array([ True]), array([False])].
The calculation took 10.1302 seconds
C7_eq results for both configurations are: [array([False]), array([ True])].
The calculation took 0.0015 seconds
# wrapping the redefined state functions works as usual
alpha_R_redef_wrapped = asynctraj.PyTrajectoryFunctionWrapper(alpha_R_redef)
C7_eq_redef_wrapped = asynctraj.PyTrajectoryFunctionWrapper(C7_eq_redef)
# and applying them to trajectories too
states = [alpha_R_redef_wrapped, C7_eq_redef_wrapped]
start = time.time()
states_for_conf_in_alphaR = await asyncio.gather(*(sf(conf_in_alphaR) for sf in states))
states_for_conf_in_C7eq = await asyncio.gather(*(sf(conf_in_C7eq) for sf in states))
end = time.time()
print(f"States (alphaR, C7_eq) for conf_in_alphaR: {states_for_conf_in_alphaR}.")
print(f"States (alphaR, C7_eq) for other_conf: {states_for_conf_in_C7eq}.")
print(f"The calculation took {round(end-start, 4)} seconds")
# Here again the second application should be slightly faster due to caching
start = time.time()
states_for_conf_in_alphaR = await asyncio.gather(*(sf(conf_in_alphaR) for sf in states))
states_for_conf_in_C7eq = await asyncio.gather(*(sf(conf_in_C7eq) for sf in states))
end = time.time()
print(f"Running the same 'calculation' again took {round(end-start, 4)} seconds...we just retrieved the results from cache once again.")
States (alphaR, C7_eq) for conf_in_alphaR: [array([ True]), array([False])].
States (alphaR, C7_eq) for other_conf: [array([False]), array([ True])].
The calculation took 0.0038 seconds
Running the same 'calculation' again took 0.0014 seconds...we just retrieved the results from cache once again.
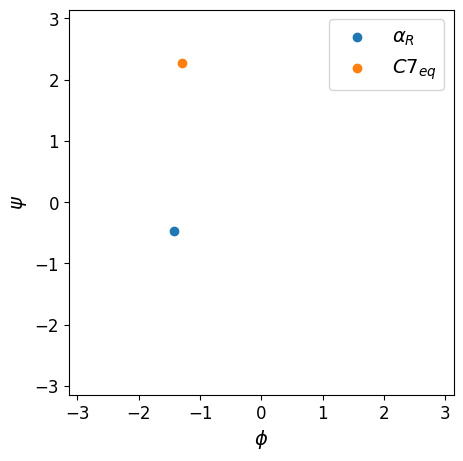
Plot the two configurations in the plane of \(\phi\) and \(\psi\)#
We will use the wrapped descriptor_func_psi_phi and plot the results.
vals_in_alphaR, vals_in_C7eq = await asyncio.gather(psi_phi_wrapped(conf_in_alphaR), psi_phi_wrapped(conf_in_C7eq))
fig, axs = plt.subplots(figsize=(5,5))
axs.scatter(vals_in_alphaR[:, 1], vals_in_alphaR[:, 0], label=r"$\alpha_R$")
axs.scatter(vals_in_C7eq[:, 1], vals_in_C7eq[:, 0], label=r"$C7_{eq}$")
axs.set_xlim(-np.pi, np.pi)
axs.set_ylim(-np.pi, np.pi)
axs.set_xlabel(r"$\phi$", size=14)
axs.set_ylabel(r"$\psi$", size=14)
axs.tick_params(labelsize=12)
axs.set_aspect("equal")
axs.legend(fontsize=14);